| MitImpact id |
MI.22186 |
MI.22187 |
MI.22185 |
| Chr |
chrM |
chrM |
chrM |
| Start |
13708 |
13708 |
13708 |
| Ref |
G |
G |
G |
| Alt |
A |
C |
T |
| Gene symbol |
MT-ND5 |
MT-ND5 |
MT-ND5 |
| Extended annotation |
mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 5 |
mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 5 |
mitochondrially encoded NADH:ubiquinone oxidoreductase core subunit 5 |
| Gene position |
1372 |
1372 |
1372 |
| Gene start |
12337 |
12337 |
12337 |
| Gene end |
14148 |
14148 |
14148 |
| Gene strand |
+ |
+ |
+ |
| Codon substitution |
GCA/ACA |
GCA/CCA |
GCA/TCA |
| AA position |
458 |
458 |
458 |
| AA ref |
A |
A |
A |
| AA alt |
T |
P |
S |
| Functional effect general |
missense |
missense |
missense |
| Functional effect detailed |
missense |
missense |
missense |
| OMIM id |
516005 |
516005 |
516005 |
| HGVS |
NC_012920.1:g.13708G>A |
NC_012920.1:g.13708G>C |
NC_012920.1:g.13708G>T |
| HGNC id |
7461 |
7461 |
7461 |
| Respiratory Chain complex |
I |
I |
I |
| Ensembl gene id |
ENSG00000198786 |
ENSG00000198786 |
ENSG00000198786 |
| Ensembl transcript id |
ENST00000361567 |
ENST00000361567 |
ENST00000361567 |
| Ensembl protein id |
ENSP00000354813 |
ENSP00000354813 |
ENSP00000354813 |
| Uniprot id |
P03915 |
P03915 |
P03915 |
| Uniprot name |
NU5M_HUMAN |
NU5M_HUMAN |
NU5M_HUMAN |
| Ncbi gene id |
4540 |
4540 |
4540 |
| Ncbi protein id |
YP_003024036.1 |
YP_003024036.1 |
YP_003024036.1 |
| PhyloP 100V |
0.633 |
0.633 |
0.633 |
| PhyloP 470Way |
-0.807 |
-0.807 |
-0.807 |
| PhastCons 100V |
0 |
0 |
0 |
| PhastCons 470Way |
0.002 |
0.002 |
0.002 |
| PolyPhen2 |
benign |
possibly_damaging |
benign |
| PolyPhen2 score |
0.02 |
0.86 |
0.4 |
| SIFT |
neutral |
neutral |
neutral |
| SIFT score |
0.48 |
0.26 |
0.5 |
| SIFT4G |
Tolerated |
Damaging |
Damaging |
| SIFT4G score |
0.078 |
0.003 |
0.004 |
| VEST |
Neutral |
Neutral |
Neutral |
| VEST pvalue |
0.37 |
0.11 |
0.37 |
| VEST FDR |
0.5 |
0.4 |
0.5 |
| Mitoclass.1 |
neutral |
damaging |
neutral |
| SNPDryad |
Neutral |
Neutral |
Neutral |
| SNPDryad score |
0.01 |
0.62 |
0.56 |
| MutationTaster |
Polymorphism |
Polymorphism |
Polymorphism |
| MutationTaster score |
1.0 |
1.0 |
1.0 |
| MutationTaster converted rankscore |
0.08975 |
0.08975 |
0.08975 |
| MutationTaster model |
simple_aae |
simple_aae |
simple_aae |
| MutationTaster AAE |
A458T |
A458P |
A458S |
| fathmm |
Tolerated |
Tolerated |
Tolerated |
| fathmm score |
1.25 |
1.21 |
1.32 |
| fathmm converted rankscore |
0.36512 |
0.37230 |
0.35219 |
| AlphaMissense |
likely_benign |
likely_pathogenic |
likely_benign |
| AlphaMissense score |
0.1315 |
0.9753 |
0.2215 |
| CADD |
Neutral |
Neutral |
Neutral |
| CADD score |
1.254751 |
2.050274 |
1.87414 |
| CADD phred |
12.03 |
16.53 |
15.43 |
| PROVEAN |
Tolerated |
Damaging |
Tolerated |
| PROVEAN score |
-1.5 |
-2.87 |
-1.51 |
| MutationAssessor |
low |
high |
high |
| MutationAssessor score |
1.6 |
4.175 |
3.83 |
| EFIN SP |
Damaging |
Damaging |
Damaging |
| EFIN SP score |
0.29 |
0.536 |
0.538 |
| EFIN HD |
Neutral |
Neutral |
Neutral |
| EFIN HD score |
0.736 |
0.396 |
0.704 |
| MLC |
Deleterious |
Deleterious |
Deleterious |
| MLC score |
0.6244191 |
0.6244191 |
0.6244191 |
| PANTHER score |
0.627 |
. |
. |
| PhD-SNP score |
0.783 |
. |
. |
| APOGEE1 |
Pathogenic |
Neutral |
Neutral |
| APOGEE1 score |
0.76 |
0.33 |
0.34 |
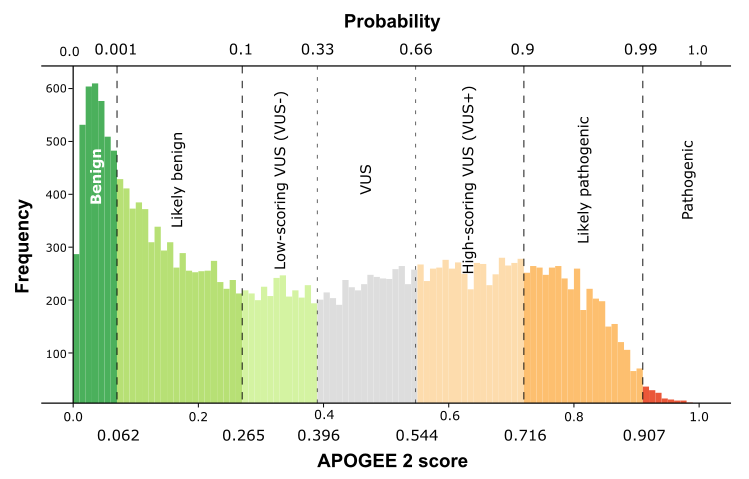
| APOGEE2 |
VUS- |
VUS+ |
Likely-benign |
| APOGEE2 score |
0.307025749980826 |
0.696841096401621 |
0.260757785070776 |
| CAROL |
neutral |
neutral |
neutral |
| CAROL score |
0.5 |
0.89 |
0.44 |
| Condel |
deleterious |
neutral |
deleterious |
| Condel score |
0.73 |
0.2 |
0.55 |
| COVEC WMV |
neutral |
deleterious |
neutral |
| COVEC WMV score |
-6 |
1 |
-3 |
| MtoolBox |
neutral |
neutral |
neutral |
| MtoolBox DS |
0.21 |
0.38 |
0.26 |
| DEOGEN2 |
Tolerated |
Tolerated |
Tolerated |
| DEOGEN2 score |
0.03024 |
0.343481 |
0.028973 |
| DEOGEN2 converted rankscore |
0.21483 |
0.71212 |
0.20865 |
| Meta-SNP |
Neutral |
. |
. |
| Meta-SNP score |
0.4 |
. |
. |
| PolyPhen2 transf |
medium impact |
low impact |
medium impact |
| PolyPhen2 transf score |
0.86 |
-1.5 |
-0.58 |
| SIFT_transf |
medium impact |
medium impact |
medium impact |
| SIFT transf score |
0.21 |
-0.02 |
0.23 |
| MutationAssessor transf |
medium impact |
high impact |
medium impact |
| MutationAssessor transf score |
0.15 |
2.13 |
1.5 |
| CHASM |
Neutral |
Neutral |
Neutral |
| CHASM pvalue |
0.68 |
0.74 |
0.72 |
| CHASM FDR |
0.85 |
0.85 |
0.85 |
| ClinVar id |
9696.0 |
. |
. |
| ClinVar Allele id |
24735.0 |
. |
. |
| ClinVar CLNDISDB |
Human_Phenotype_Ontology:HP:0001086,Human_Phenotype_Ontology:HP:0001112,MONDO:MONDO:0010788,MedGen:C0917796,OMIM:535000,Orphanet:104|MONDO:MONDO:0009723,MedGen:C0023264,OMIM:256000,Orphanet:506 |
. |
. |
| ClinVar CLNDN |
Leber_optic_atrophy|Leigh_syndrome |
. |
. |
| ClinVar CLNSIG |
Benign |
. |
. |
| MITOMAP Disease Clinical info |
LHON / Increased MS risk / higher freq in PD-ADS |
. |
. |
| MITOMAP Disease Status |
Conflicting reports |
. |
. |
| MITOMAP Disease Hom/Het |
+/+ |
./. |
./. |
| MITOMAP General GenBank Freq |
7.459% |
. |
. |
| MITOMAP General GenBank Seqs |
4560 |
. |
. |
| MITOMAP General Curated refs |
15470367;18619472;10520236;1634041;19220304;27119776;19005266;16044424;1550131;18545700;10545708;1417830;17603766;21067478;18205894;18590963;8163275;16901986;18806273;18712405;18308428;18270557;15234467;21288980;1900003;8680405;15060117;24002810;20691156;11179019;21457906;30369864;21694444;19370763;16313983;9027481;15625560;16532388;11349229;11935318;21878127;1732158;7710535;8600429;9561330;10216058;34573281;19527690;12888043;9302261;19223931;8053461;7599218;17003408;20304802;29486301;36322731;12150954;7763260;31798871;9150158;7942855;31797714;20067846;21978175;18477584;15382008;22561905;32094358;7814218;7977345;7901141;19026397;17406640;15972314;19151382;11571560;17660050;9915963;8213820;16404693;18775412;19062322;31152278;15286228;7635294;18286226;16773565;18931934;19489744;10737123;7770132;19427920;21724059;10424809;28341142;12618962;16960846;8978068;25313049;18668590;18322915;18853457;11820805;18691441;32887465;1463007;19130794;19818876;27498855;8741876;15591266;23304069;11938495;16714301;8071952;10680807;12802679;24069186;15932126;15975594;10234520;16050984;19500771;18386806;21041797;19340307;8024249;12937995;18810306;29987491;10936107;8755941;1764087;11339587 |
. |
. |
| MITOMAP Variant Class |
polymorphism;disease |
. |
. |
| gnomAD 3.1 AN |
56361.0 |
. |
. |
| gnomAD 3.1 AC Homo |
4263.0 |
. |
. |
| gnomAD 3.1 AF Hom |
0.0756374 |
. |
. |
| gnomAD 3.1 AC Het |
21.0 |
. |
. |
| gnomAD 3.1 AF Het |
0.000372598 |
. |
. |
| gnomAD 3.1 filter |
PASS |
. |
. |
| HelixMTdb AC Hom |
19999.0 |
. |
. |
| HelixMTdb AF Hom |
0.10204457 |
. |
. |
| HelixMTdb AC Het |
86.0 |
. |
. |
| HelixMTdb AF Het |
0.00043881356 |
. |
. |
| HelixMTdb mean ARF |
0.51484 |
. |
. |
| HelixMTdb max ARF |
0.96842 |
. |
. |
| ToMMo 54KJPN AC |
1215 |
. |
. |
| ToMMo 54KJPN AF |
0.022375 |
. |
. |
| ToMMo 54KJPN AN |
54302 |
. |
. |
| COSMIC 90 |
. |
. |
. |
| dbSNP 156 id |
rs28359178 |
. |
. |